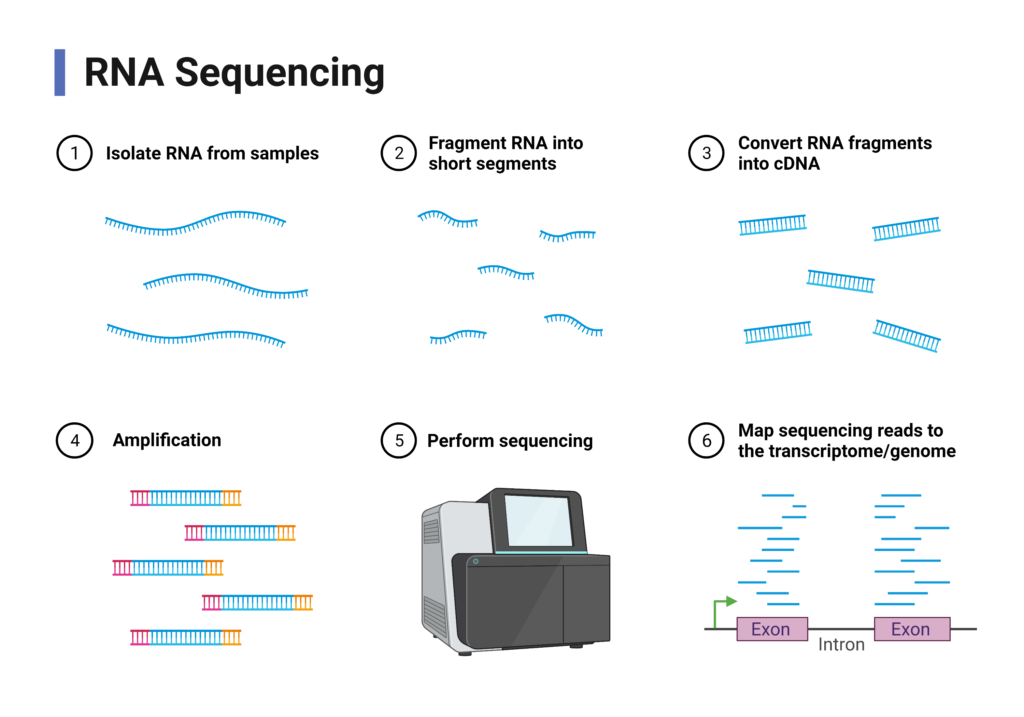
High-throughput sequencing methods have allowed transcriptomics [1] to develop significantly over the past decade. The ability to in-depth study of complete transcriptomes using RNA sequencing (RNA-seq) has contributed to many important discoveries and has now become the standard method in biomedical research.
What is RNA-seq?
RNA-seq is a technique that allows the analysis of the entire transcriptome with high efficiency. It allows, among others the characteristics of alternative splicing products [2] or the detection of gene fusion products [3]. Accurate knowledge of the transcriptome is important for understanding, among others, the mechanisms of development and growth of cells, and thus of whole organisms. Thanks to the analysis of the RNA sequence, we can study which genes are activated or turned off in a given cell.
Unfortunately, RNA-seq techniques are often performed in large numbers and the results represent patterns of gene expression are averaged for hundreds or millions of cells, which can mask biologically significant changes in cells.
How to overcome this?
Single-cell RNA sequencing (scRNA-srq) is one method of solving this problem. This method enables the assessment of the basic biological characteristics of cell populations and biological systems with unprecedented precision by isolating single cells, collecting their transcripts and creating sequencing libraries, in which the transcripts are mapped to individual cells. ScRNA-srq therefore allows an RNA molecule to be characterized in high resolution and on a large scale by comparing the transcriptomes of single cells within a population. Transcriptional differences between individual cells can be used, for example, to identify rare populations of cells that would otherwise go undetected.
Although different scRNA-seq methods have been investigated and their strengths and weaknesses have been identified, scientists still need to choose between methods with high cell throughput, but low transcript coverage, and methods with high sensitivity and full-length transcript coverage. Therefore, based on the Smart-seq3 method, which offers the highest sensitivity, it detects the largest number of genes in the cell, the Smart-seq3xpress technique was created.
New quality of sensitivity – I.DOT
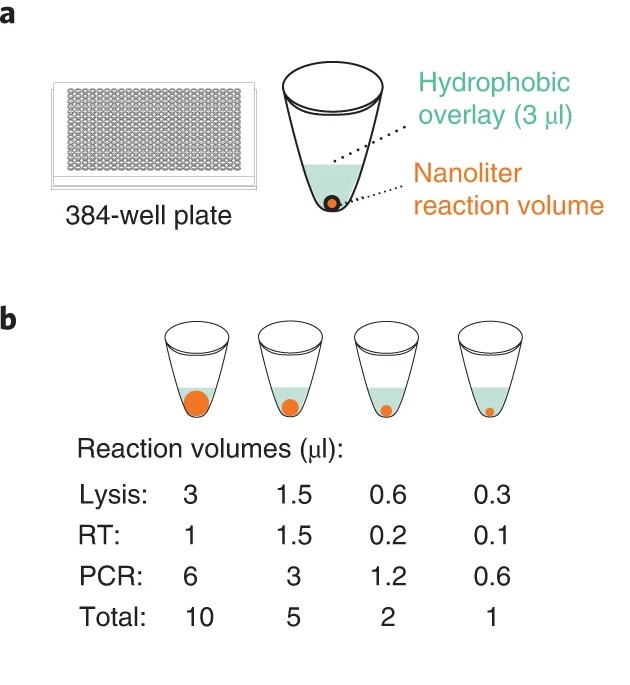
This technique was created thanks to the I.DOT device by the Dispendix company. A precise, low-volume, non-contact fluid dispenser with a droplet volume of only a few nanoliters. It enhances the Smart-seq3 process by minimizing the amount of reagents used and increasing cellular throughput without compromising data quality. By excluding several time- and resource- consuming experimental stages, the duration of the experiment was shortened to one working day.
Find out more about I.DOT and dispensers!
Using the I.DOT dispenser, the authors were able to reduce the reaction volumes of many steps by half, or even to 1/10 of the previous amounts, and assess these conditions on the cells of human cell lines: K562 – myeloid leukemia cell line and HEK293FT – embryonic kidney cell line . As a result, costs and valuable experiment duration were drastically reduced, and the use of plastic consumables was minimized.
With the development of Smart-seq3xpress, high-sensitivity scRNA-seq with a resolution suitable for exploring new cell types in human tissues and for building a large-scale cell atlas can be performed for the first time.
[1] Transcriptomics – a branch of biological sciences that studies the transcriptome, which is the set of mRNA molecules in a cell, at a given moment.
[2] Alternative splicing – joining coding genes together in different ways. It is the source of protein variability.
[3] Gene fusion – combining fragments of 2 genes, thanks to it a fusion gene is created.
Source: Hagemann-Jensen, M., Ziegenhain, C. & Sandberg, R. Scalable single-cell RNA sequencing from full transcripts with Smart-seq3xpress. Nat Biotechnol (2022). https://doi.org/10.1038/s41587-022-01311-4





