Wysokowydajne metody sekwencjonowania umożliwiły znaczny rozwój transkryptomiki [1] w ciągu ostatniej dekady. Możliwość dogłębnego badania pełnych transkryptomów za pomocą sekwencjonowania RNA (RNA-seq) przyczyniła się do wielu ważnych odkryć i stała się obecnie standardową metodą w badaniach biomedycznych.
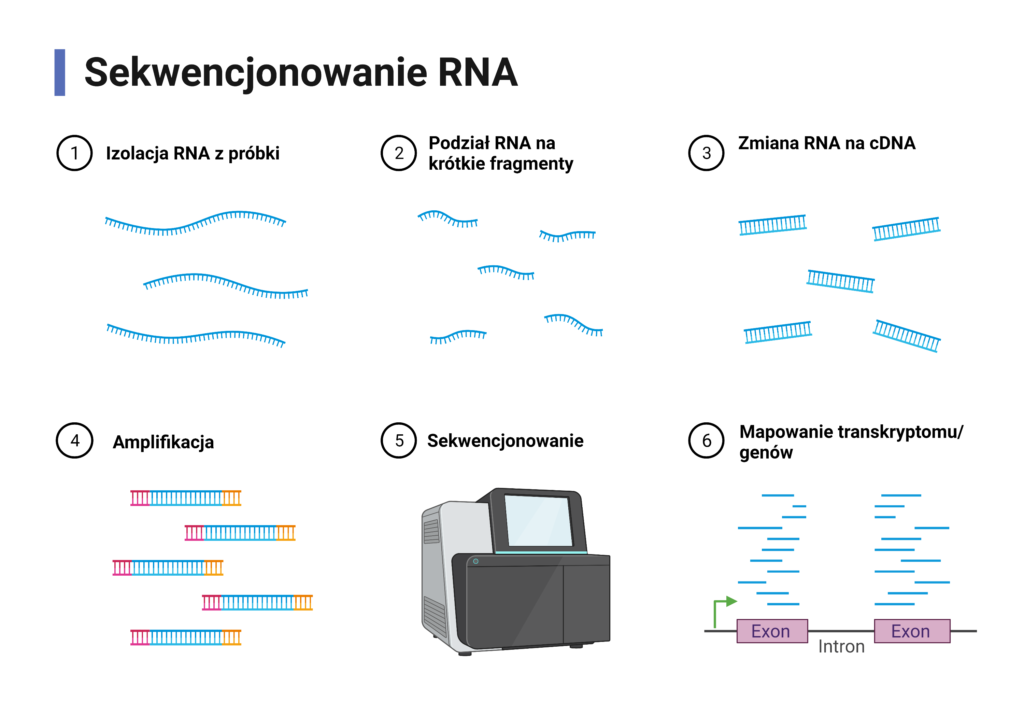
Czym jest RNA-seq?
RNA-seq jest techniką pozwalającą na analizę całego transkryptomu z wysoką wydajnością. Pozwala na charakterystykę produktów alternatywnego składania [2] czy wykrycie produktów fuzji genów [3]. Dokładne poznanie transkryptomu jest istotne dla zrozumienia m.in mechanizmów rozwoju i wzrostu komórek, a co za tym idzie, całych organizmów. Dzięki analizie sekwencji RNA możemy badać, które geny są aktywowane lub wyłączone w danej komórce.
Niestety techniki RNA-seq często wykonywane są w dużych ilościach, a wyniki reprezentujące wzorce ekspresji genów są uśrednione dla setek czy milionów komórek, co może maskować biologicznie znaczące zmiany w komórkach.
Jak pokonać tę przeszkodę?
Sekwencjonowanie RNA na poziomie pojedynczej komórki (scRNA-srq) jest jedną z metod rozwiązywania tego problemu. Metoda ta umożliwia ocenę podstawowych cech biologicznych populacji komórek i układów biologicznych z niespotykaną dotąd precyzją poprzez izolację pojedynczych komórek, zbieranie ich transkryptów i tworzenie bibliotek sekwencjonowania, w których transkrypty są mapowane na poszczególne komórki. ScRNA-srq pozwala więc na opisanie cząsteczki RNA z wysoką rozdzielczością i na dużą skalę, porównując transkryptomy pojedynczych komórek w obrębie populacji. Różnice transkrypcyjne między poszczególnymi komórkami mogą zostać wykorzystane np. do identyfikacji rzadkich populacji komórek, które w innym przypadku pozostałyby niewykryte.
Chociaż zbadano różne metody scRNA-seq oraz zidentyfikowano ich mocne i słabe strony, naukowcy wciąż muszą wybierać pomiędzy metodami o wysokiej przepustowości komórek, ale niskim pokryciem transkryptu, a metodami z wysoką czułością i pokryciem transkryptu pełnej długości. Dlatego w oparciu o metodę Smart-seq3, która oferuje najwyższą czułość (wykrywa największą ilość genów w komórce) powstała technika Smart-seq3xpress.
Nowa jakość czułości – I.DOT
Technika ta powstała dzięki urządzeniu I.DOT firmy Dispendix – dokładnemu, bezdotykowemu dyspenserowi płynów o małej objętości, którego objętość kropli może wynosić tylko kilka nanolitrów. Usprawnia on proces Smart-seq3 poprzez minimalizację ilości użytych odczynników i zwiększenie przepustowości komórkowej bez pogorszenia jakości danych. Poprzez wykluczenie kilku czaso- i zasobochłonnych etapów eksperymentalnych skrócono czas trwania eksperymentu do jednego dnia roboczego.
Dowiedz się więcej o I.DOT i dyspenserach!
Za pomocą dyspensera I.DOT autorzy pracy byli w stanie zredukować objętości reakcji wielu etapów o połowę, a nawet do 1/10 wcześniejszych ilości i ocenić te warunki na komórkach ludzkich linii komórkowych: K562- linii komórek białaczki szpikowej i HEK293FT- linii komórek embrionalnych nerki. W rezultacie radykalnie obniżono koszty i cenny czas trwania eksperymentu oraz zminimalizowano zużycie materiałów eksploatacyjnych z tworzyw sztucznych.
Wraz z rozwojem Smart-seq3xpress po raz pierwszy można przeprowadzić wysokiej czułości scRNA-seq z rozdzielczością odpowiednią do poznawania nowych typów komórek w tkankach ludzkich oraz do budowy atlasu komórkowego na dużą skalę.
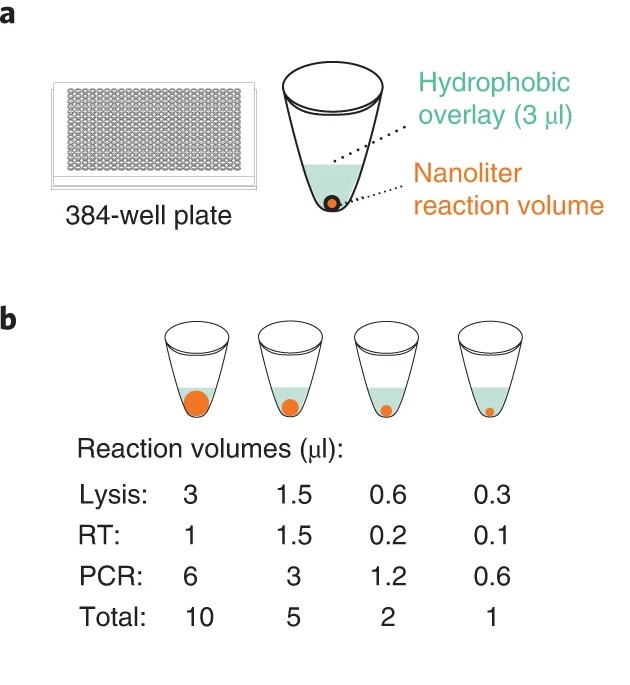
Źródło: https://www.nature.com/articles/s41587-022-01311-4/figures/1
[1] Transkryptomika – dziedzina nauk biologicznych zajmująca się badaniem transkryptomu, czyli zestawu cząsteczek mRNA w komórce w danym momencie.
[2] Alternatywne składanie – łączenie ze sobą genów kodujących na różne sposoby. Jest ono źródłem zmienności białek.
[3] Fuzja genów – łączenie fragmentów dwóch genów, dzięki czemu powstaje gen fuzyjny.
Źródło: Hagemann-Jensen, M., Ziegenhain, C. & Sandberg, R. Scalable single-cell RNA sequencing from full transcripts with Smart-seq3xpress. Nat Biotechnol (2022). https://doi.org/10.1038/s41587-022-01311-4





